
As pharmaceutical consultants, the scientists and regulatory professionals at PDG® design pharmacokinetic (PK) protocols for a wide variety of applications. This paper has been written to provide the reader with a basic review of pharmacokinetic and pharmacodynamics (PD) principles as well as an exposure to a cross section of the utility of PK studies. While not meant as a comprehensive compendium, included are brief overviews of PK/PD principles and discussions of the following topics as they relate to PK:
- PILOT STUDIES
- ANDAs/505(b)(2)
- BIOWAIVERS
- BIOSIMILARS
- BRIDGING
- SAFETY
- DOSE RESPONSE/DOSE REGIMEN
- ROUTE OF ADMINISTRATION
- FOOD/FASTING EFFECTS
- PEDIATRIC CONSIDERATIONS
- GERIATRIC CONSIDERATIONS
PK is the study of how the body processes a drug. There are several biologic factors that are affected by drugs, commonly referred to as “ADME”[1], [2]:
A – Absorption – how fast and how completely the drug is absorbed into the body
D – Distribution – how the drug is distributed through the various body tissues
M – Metabolism – how and to what extent the drug is metabolized (chemically broken down into other molecules, i.e. metabolites)
E – Excretion – how rapidly the drug and its metabolites are eliminated from the body.
In addition to ADME, other patient specific factors impact the way drugs are processed such as genetic make-up, age, gender, or influences of the disease state. Safe & effective use of any drug is dependent upon delivery of an adequate amount (concentration) of the drug to the receptor sites in a timely manner (rate) to properly influence the bodily systems in need of the drug. Hence, the science of PK is the application of quantification and modeling techniques to the timing of drug ADME.[3]
By graphing drug concentration versus time, estimates of a drug’s PK properties may be derived, resulting in a plasma drug concentration-time curve. The area under this curve (AUC) is a reflection of the body’s actual exposure to drug after administration of a dose expressed in mg*h/L (see Figure 1).[4]
Figure 1
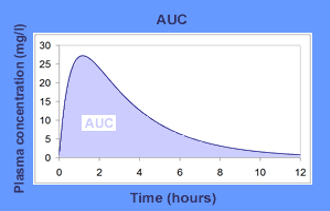
Area under the plasma drug concentration-time curve example. Adapted from Pharmacokinetics, Thierry Buclin, et. al.
Three conceptual PK parameters are used to describe the outcomes of the ADME factors in a quantitative manner:[5]
- Clearance – rate of drug elimination divided by the plasma concentration, yielding units of flow ml/min that can be corrected for bodyweight (ml/min/kg); thus expressing the body’s ability to eliminate the drug by either metabolism or excretion. Clearance ultimately determines the dose rate needed to maintain the desired plasma concentration.
- Bioavailability – proportion of dose reaching systematic circulation, usually expressed in percentage (%). IV drugs score 100%. All other routes of administration are diminished by incomplete absorption or metabolism and should always be less than 100%.
- Volume of Distribution – amount of the drug in the body divided by the plasma concentration expressed in volume units that may be corrected for bodyweight, e.g. L/kg.
PK studies are commonly used for Phase 1 testing to determine initial dose regimens, or when the drug reaches steady state. As data is collected and analyzed, the sponsor can refine the dose regimens for later phase studies to ensure they are safe and effective for use. Regulatory authorities rely upon PK data to assess the safety and efficacy of the drugs and to ensure the testing and calculations have been done accurately by the sponsors. They may compare the labeled dosage and administration claims to study results to ensure dose adjustments are not needed for food effects or special populations (elderly, renal impaired, etc.).[6]
PHARMACOKINETICS vs. PHARMACODYNAMICS
While PK is the study of how the body processes a drug, PD is the study of the effect the drug has on the body.[7] PD data is used to assess whether the actions of the drug are as intended (i.e. is the drug effective?). Many Phase I studies also include a PD component.[8] This has become more prevalent as regulatory standards increase in rigor, clinical trials take longer, and costs continue to rise. Such factors justify sponsor assessments of therapeutic potential and drug safety as early and as comprehensively as possible. Drug effects are measured in several ways, including:
- Analyses of receptors, either targeted or not
- Indirect alteration of endogenous agonist or antagonist activity
- Inhibition of transport processes
- Alteration of enzyme activity
- Activities of an analogue of natural ligand (e.g. replacement therapy)[9]
As with PK, each method used to measure the PD response to drug administration must be quantifiable. This can prove difficult, especially in Phase 1 studies conducted on healthy volunteers.[10] However, both disciplines are essential in the evaluation of the interaction between a drug and humans, and each plays a critical role in ensuring safe and effective use.
PK/PD MODELING
PK/PD modeling consists of characterizing the relationship between PK (concentration vs. time) and PD (effect vs. time). Deployment of PK/PD modeling at the earliest stages of research may facilitate a more successful transition to drug development.[11] Elucidation of the PK/PD relationship contributes to understanding the mechanism of action of the drug and may improve compound design. PK/PD modeling can also enhance the extrapolation of in vitro potency data to the in vivo setting, thereby reducing the number of animal studies and improving the application of animal findings to the clinical program. Such outputs have the potential of shortening development time by more accurately predicting/estimating therapeutic index dose ranges in early clinical testing.[12] There are also PK/PD modeling methods that are appropriate for applied therapeutics.[13]
1. PILOT STUDIES
Regardless of the data need, the possibility of a pilot study is always an important consideration. Generally speaking, PK studies afford the earliest look at data in humans. The pilot PK study serves as an “early look at the early look” and has the potential of saving time and money, as well as informing pivotal studies. As noted in the safety section of this paper below, pilot studies also have a place in population pharmacokinetics.[14] The following examples of pilot study utility are excepted directly from FDA bioequivalence regulatory guidances. However, the applicability is universal:
If the sponsor chooses, a pilot study in a small number of subjects can be carried out before proceeding with a full BE study. The study can be used to validate analytical methodology, assess variability, optimize sample collection time intervals, and provide other information. For example, for conventional immediate-release products, careful timing of initial samples may avoid a subsequent finding in a full-scale study that the first sample collection occurs after the plasma concentration peak. For modified-release products, a pilot study can help determine the sampling schedule to assess lag time and dose dumping.[15]
2. ANDAs/505(b)(2)
PK studies are perhaps best known as those relied upon to prove bioequivalence to reference listed drugs (RLDs) for generic (505(j) or ANDA) and 505(b)(2) new drug applications. ANDA approvals require evidence that the generic drug product has the same active ingredient, dosage form, strength, route of administration and conditions of use and that the proposed drug product is bioequivalent to the reference listed drug.[16], [17] On the other hand, 505(b)(2) applications seek approval based on differentiated conditions of use. However, 505(b)(2) applications must still prove bioequivalence (or at least as bioavailable) to the RLD and may also require clinical studies.[18]
For drugs that are not intended to be absorbed into the bloodstream, “FDA may establish alternative methods to show bioequivalence that may be expected to detect a significant difference between the drug and the listed drug in safety and therapeutic effect.”[19] In June 2010, FDA published a guidance describing their processes for publishing product-by-product guidance on the design of BE studies to support ANDAs. This guidance, and subsequent product-specific guidances, marked the end of the previous approach of individual applicant requests for such information.[20] FDA now adds to, and updates these guidances periodically.[21] As of this writing (January 2017) there were at least 1573 bioequivalence recommendations for specific products, with no less than sixty-seven of them added or revised during the previous three months.[22]
3. BIOWAIVERS
Anyone submitting an NDA, (including ANDA) may request that FDA waive the requirement of “evidence measuring the in vivo bioavailability or demonstrating the in vivo bioequivalence of the drug product that is the subject of the application.” Underlying circumstances of waiver requests are many and varied. For example, the “in vivo bioavailability or bioequivalence of the drug product may be self-evident”.[23] This is generally applicable to (among others) injectables, ophthalmic or otic solutions, inhaled gases, topical solutions, nasal solutions, that contain an active in the same concentration and dosage form as a drug product that is the subject of an approved NDA/ANDA. Formulation changes that significantly affect absorption or local availability (for products intended to act locally) will generally rule out a biowaiver.[24]
Certain old drugs (e.g. Drug Efficacy Study Implementation (DESI) products) may qualify for biowaivers,[25] and FDA may waive BE requirements for protection of the public health.[26] There are also cases in which FDA may allow BA or BE to be demonstrated by in vitro in lieu of in vivo data. This generally applies to drugs of a same dosage form, different strength, and proportionally similar active and inactive ingredients to other sponsor approved drugs. BA of the other drug products will have been measured, both products are subject of in vitro testing approved by FDA, or there is evidence in the NDA that the in vitro data has been correlated with in vivo data. Delayed or extended release dosage forms generally do not qualify.[27]
Biowaivers may also be requested for immediate release solid oral dosage forms using an approach termed the Biopharmaceutics Classification System (BCS). The BCS is described in the guidance entitled Waiver of In-Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System and is a “scientific framework for classifying drug substances based on their aqueous solubility and intestinal permeability”.[28]
4. BIOSIMILARS
Made from living organisms, biologics are relatively large and complex molecules compared to conventional chemical drugs. Biologics tend to be made up of proteins (and/or constituent amino acids), carbohydrates (e.g. sugars), nucleic acids (e.g. DNA), or combinations thereof, and also include cells or tissues used in transplantation. Because of their relatively complex chemical structure and method of manufacture, biosimilars (aka follow-on biologics) are never identical to the brand-name product, but will instead be shown to be highly similar. Nor are they developed using ANDA or 505(b)(2) submissions. However, we have included the discussion in this paper because of similar requirements shared with ANDAs and 505(b)(2)s.
In April 2006, the European Medicines Agency (EMA) approved Europe’s first ever biosimilar, Omnitrope®, a human growth hormone. Since that time, the EMA has authorized a total of 20 biosimilars. FDA also approved Omnitrope® in 2006, but it followed an April 2006 court ruling that the agency must review the application. In doing so, FDA reviewed the drug as a 505(b)(2), stated that the approval did not establish a pathway for approval of other follow-on biologics and that Congress would need to change the law in order for the agency to continue doing so.[29]
On March 23, 2010, the President signed the Biologics Price Competition and Innovation Act of 2009 (BPCI Act) into law, which amended the Public Health Service Act (PHS Act) and created an abbreviated review pathway for biosimilars. The legislation required that sponsors submit a 351(k) Biologics License Application (BLA) that includes, inter alia, evidence that the compound is interchangeable with an already approved biological product. Approval will generally be based on the following:
Analytical studies demonstrating that the biological product is “highly similar” to the reference product notwithstanding minor differences in clinically inactive components; Animal studies (including the assessment of toxicity); and a clinical study or studies (including the assessment of immunogenicity and pharmacokinetics (PK) or pharmacodynamics (PD)) sufficient to demonstrate safety, purity, and potency in 1 or more appropriate conditions of use for which the reference product is licensed and for which licensure is sought for the biosimilar product.[30]
It is important to note that FDA maintains discretion to determine that any of the elements described above may be unnecessary in a 351(k) BLA.
FDA also “encourages the use of novel methods and study designs (e.g., endpoints, populations) to increase sensitivity to detect potential differences to support a demonstration of biosimilarity” and has stated that biosimilar development programs may be very different from those of the reference product “e.g., PK/PD in normal volunteers rather than Phase 3 clinical trial.”[31]
Therefore, FDA encourages sponsors to contact their review division early to gain input on proposed development programs.[32]
As of this writing, FDA has approved three biosimilars for marketing in the United States: Zarxio® (filgrastim-sndz) in March 2015, Inflectra® (infliximab-dyyb) in April 2016, and Erelzi™, (etanercept-szzs) in August 2016.[33]
5. BRIDGING
PK studies are useful in the development of bridging data. Bridging is narrowly defined by ICH Guidance as “a study performed in the new region to provide pharmacodynamic or clinical data on efficacy, safety, dosage and dose regimen in the new region that will allow extrapolation of the foreign clinical data to the population in the new region.”[34] For example, Epogen® (epoetin alfa) was developed by Amgen and approved by FDA in 1989 for the treatment of anemia in patients with chronic liver failure.[35] The drug went off patent in Europe in 2004.[36]
Sandoz developed its own version (HX575) which was approved as a biosimilar in Europe in 2007. The tradename of HX575 is Binocrit®, which relied upon Erypo®/Eprex® (the European formulation) as reference product. Sandoz went on to compare the US, European and HX575 formulations as follows: “The study was conducted in two parts: part A compared the European-marketed HX575 and the US-marketed Epogen®; part B compared the European-marketed Erypo®/Eprex® and HX575 manufactured at two different drug substance production sites (HX575-TT denoting the already-approved technology-transfer product from an additional manufacturing site). Bioequivalence was shown in all pair-wise comparisons.” Thus, the bioequivalence and pharmacodynamic equivalence allowed bridging of data from clinical studies using either US- or European epoetin alfa products. Because of the similarity of all three products and despite the different formulations clinical study data produced with a product sourced in either region could be used to support further marketing authorizations, including in other geographical markets.[37]
In practical use bridging is not confined to geographic differences. In a more contemporary example, on July 20, 2015, Adapt Pharma submitted a 505(b)(2) for Narcan® (naloxone hydrochloride), which proposed a change in route of administration, from injectable to intranasal. Narcan® is indicated for emergency treatment of known or suspected opioid overdose. The application was approved four months after submission, on November 18, 2015 and was launched in early 2016.[38] Because marketing of injectable Narcan® had been discontinued, the Applicant referenced a generic product, International Medicinal System’s naloxone HCl injection USP pre-filled syringe (ANDA 072076) and relied upon a relative BA study to create a scientific bridge to FDA’s previous findings for Narcan®.[39]
6. SAFETY
Differences in drug safety and efficacy among population subgroups based on varied ADME parameters are well known. PK studies during the premarket stage of a drug serve as important risk assessment tools, identifying proper dosing, drug-drug interactions and clinical situations of concern such as such as impaired liver or kidney function. Also identified are vulnerable sub-populations such as geriatric or pediatric.
Population pharmacokinetics is the science of identifying measurable factors that effect changes in the dose-concentration relationship, as well as the extent of these changes between subgroups. Changes that are associated with clinically significant shifts in the therapeutic index may inform modifications in labeled dosing regimens. The guidance on population pharmacokinetics includes labeling examples, one of which is excerpted below. In this example, “both gender and body weight influence the total drug clearance (CL) while the variability in volume of distribution (Vd) can be explained by the contribution from body weight”:
The influence of gender and body weight on the pharmacokinetics of drug X was studied using population PK analysis in 232 males (weight 52 to 138 kg) and 288 females (weight 49 to 116 kg) patients who underwent clinical therapy with drug X. The observed large inter-subject variabilities in CL and Vd could be explained by gender and body weight as follows:
CL (ml/min) = 19.3 x (Weight (kg)/75)2.55 (For males)
CL (ml/min) = 12.1 x (Weight (kg)/65)2.75 (For females)
And
Vd (L) = 12 + 0.5 x Weight (kg) (for both genders).[40]
Population modeling is most likely to be useful when the population for which the drug is intended is expected to be substantially heterogeneous or the target concentration window may be relatively narrow. Such studies are generally executed during Phase 1 and late Phase 2b of clinical drug development[41] and should be discussed with FDA at early development stage.
For example, Reeves et al., conducted a population study on the antipsychotic drug amisulpride to determine the effect on older patients, who were/are at greater risk of developing adverse reactions. The design was an off-label, observational study in older adults with Alzheimer’s disease, using a lower dose of amisulpride than administered in the original Phase 1 study in healthy “older” adults. Results were scaled based on body weight and age. Drug clearance between those aged 65 and those aged 85 was significantly different, and provided evidence that this (as well as weight) should be a factor in prescribing.[42]
7. DOSE RESPONSE/DOSE REGIMEN
Dose-response investigations utilize PK/PD “to determine the required dose and frequency to be given for a drug in a population.” Regardless of how a drug effect occurs, the concentration of the drug delivered to the receptor site controls the effect. One might hypothesize that a patient given a higher dose would yield a higher concentration of drug at the receptor sites, which in turn, would result in a greater effect. However, this is not always the case. PK studies aid sponsors in overcoming this via dose-response curves, each associated with drugs studied under identical conditions. The result is a comparison of the pharmacologic aspects of the drugs, with the dose on the x-axis and the effect (response) on the y-axis. It is important to note, a drug effect is not only a function of dose but time, and the comparison graph below (Figure 2) depicts the dose-response relationships independent of time.[43]
Figure 2 Graph: Comparison of dose-response curves
“Drug X has greater biologic activity per dosing equivalent and is thus more potent than drug Y or Z. Drugs X and Z have equal efficacy, indicated by their maximal attainable response (ceiling effect). Drug Y is more potent than drug Z, but its maximal efficacy is lower.”
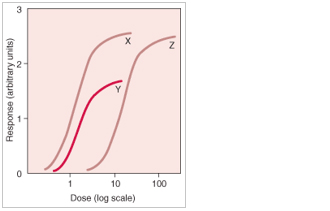
Graph example: Dose-Response Relationships. Adapted from Merck Manual Professional Version, Dose-Response Relationships.
Guptill et al., provides an example of dose response in the context of new drug development. Following preclinical studies, the authors demonstrated that their compound, CN-105, effectively downregulated the inflammatory response in acute brain injury, including spontaneous intracranial hemorrhage, and conducted a first-in-human single escalating dose and multiple dose placebo-controlled clinical trial. The objective was to show safety and illustrate the PK of CN-105. “A total of 48 subjects (12 control, 36 active) were randomized in this study; all subjects completed the study. No significant safety issues were identified with both dosing regimens, and PK analysis revealed linearity without significant drug accumulation. The median half-life in the terminal elimination phase of CN-105 following a single or repeated dosing regimen did not change (approximately 3.6 hours). With the PK and preliminary safety of CN-105 established, the drug is now poised to begin first-in-disease phase 2 clinical trials in patients with ICH who urgently need new therapeutic options.”[44]
While Guptill demonstrated the use of PK studies to establish the dosing regimen during development, PK study results may also be used for changing the dosing schedule of already approved drugs.[45]
For example, researchers at the Walter Reed Army Institute of Research recently published the results of a “phase II study which demonstrated that by changing the dosing regimen, the efficacy of malaria vaccine candidate, RTS,S/AS01, was improved to approximately 87%, compared with 63% using the current standard regimen.” Although the approved dosing regimen of the vaccine was considered effective, researchers sought improvement. The result was a regimen that specifically reduced and delayed the third dose, as well as any subsequent booster doses. Although the study did not provide enough data to implement labeling changes, one of the study’s co-authors stated that, “With these results in hand, we are planning additional studies in the United States and Africa that will seek to further refine the dosing and schedule for maximum impact.”[46]
8. ROUTE OF ADMINISTRATION
The Drugs@FDA Glossary of Terms defines route of administration as “a way of administering a drug to a site in a patient.”[47] Not to be confused with dosage form (e.g. tablets, capsules; each part of an oral route), a comprehensive list of all specific routes of administration may be found in the CDER Data Standards Manual.[48] While the manual currently lists well over one hundred routes, among those more commonly known are enteral (includes oral, sublingual, rectal), parenteral (includes subcutaneous, intramuscular, intravenous), topical, transdermal, inhalation, and intranasal.[49] Repurposing old drugs into new ones by changing the route of administration may result in patient benefits and often requires only PK studies.
In order to produce the desired clinical effect, a drug must reach its target site, whether administered on or in the body. The amount and rate at which the drug reaches the target site are in large part dependent on the route of administration.[50] As noted above, BA is the fraction of unchanged drug that makes it into systemic circulation and is useful in comparing differing routes of administration.[51] It has been said that “…the same molecule, taken at the same dose, by the same patient, but under a different administration route or a different dosage form…will not have the same kinetics of absorption, nor the same bioavailability.”[52] Herein lies the opportunity associated with repurposing old drugs through the development of new routes of administration.
When deciding upon development of a new route of administration, two major factors to consider are therapeutic concerns and drug properties. Therapeutic concerns relate to variables such as accessibility of the target site, desired onset rate, duration of effect and patient compliance. Critical drug property concerns include the plasma concentration-time profile and physicochemical characteristics, such as molecular size, ionization status and lipid solubility. Table 1 illustrates advantages and disadvantages of certain common routes of administration.”[53]
Table 1. Advantages and disadvantages of some common routes of drug administration
[wpsm_comparison_table id=”23″ class=”customTable threeColumn”]
Adapted from Drug Administration, Debra Si Mui Sim
9. FOOD/FASTING EFFECTS
Fed and fasting conditions are among the various factors that impact the way drugs are processed in the body. According to the 2002 Food-Effect Guidance:
Food can change the BA of a drug and can influence the BE between test and reference products.
Food effects on BA can have clinically significant consequences. Food can alter BA by various
means, including:
- Delay gastric emptying
- Stimulate bile flow
- Change gastrointestinal (GI) pH
- Increase splanchnic blood flow
- Change luminal metabolism of a drug substance
- Physically or chemically interact with a dosage form or a drug substance
Food effects on BA are generally greatest when the drug product is administered shortly after a meal is ingested. The nutrient and caloric contents of the meal, the meal volume, and the meal temperature can cause physiological changes in the GI tract in a way that affects drug product transit time, luminal dissolution, drug permeability, and systemic availability. In general, meals that are high in total calories and fat content are more likely to affect the GI physiology and thereby result in a larger effect on the BA of a drug substance or drug product. We recommend use of high-calorie and high-fat meals during food-effect BA and fed BE studies.[54]
When planning BA/BE studies it is important to note that there are separate guidances with regard to fed/fasting PK designs for ANDAs[55] and for INDs/NDAs.[56]
As noted above FDA has published over 1500 product specific guidances. Fed BE and food effect BA are both utilized in the investigational phase of new drug research. BA studies are typically undertaken as Phase 1 during the IND period in order to assess the effects of food on the rate and extent of absorption. In contrast, fed BE studies, are generally conducted as part of ANDA development to demonstrate BE to the RLD under fed conditions. Fed BE studies may be conducted for new drugs as well, if data regarding a possible food effect is required.[57]
According to the guidance, food-effect BA studies may be exploratory and descriptive, or may be used to substantiate a label claim. The following are among the exposure measures and PK parameters that should be obtained from the resulting concentration-time curves for both the test and reference products in food-effect BA and fed BE studies:[58]
- Total exposure, or area under the concentration-time curve (AUC0-inf, AUC0-t)
- Peak exposure (C max)
- Time to peak exposure (T max)
- Lag-time (tlag) for modified-release products, if present
- Terminal elimination half-life
- Other relevant pharmacokinetic parameters
10. PEDIATRIC CONSIDERATIONS
It is well known that the physiologic factors observed with pediatric studies affect ADME very differently than in adults. Children’s dynamic and developing physiology includes “factors such as gastric pH, intestinal transit time, immaturity of secretion and activity of bile and pancreatic fluid among other factors determine the oral bioavailability of pediatric and adult populations.”[59] As children grow and develop, so to do metabolizing enzymes, renal, and liver functions, each of which may affect drug disposition.
The differences between pediatric and adult dosing are not straightforward. There are substantial differences that depend on a child’s age/stage of body development, that justify sub-classifications: neonatal, infant, child and adolescent. Children have increased absorption and retention (e.g. lead), slower elimination in the newborn state and less P450 metabolism before 6 months of age. Therefore, grouping pediatrics together as one population or studying them as small adults, using extrapolation from adult data to make dosing determinations is generally not appropriate. More extensive PK studies are needed to ensure the safety of drug dosages in children, and FDA often specifies the age ranges for testing in children with specific drug products.[60]
Illustrative of the differences between FDA approved safe pediatric and adult usage, the following table references various drugs and their pediatric approval status.
Table 2. Pediatric indication for some drugs by the FDA.[61]
[wpsm_comparison_table id=”25″ class=”customTable sixColumn”]
![]() Approved for pediatric use from birth.
Approved for pediatric use from birth. ![]() Not approved for pediatric use.
Not approved for pediatric use.
Adapted from Factors and Mechanisms for Pharmacokinetic Differences between Pediatric Population and Adults, Eva Fernandez et. al.
The Federal Food, Drug, and Cosmetic Act was amended in 2003 to authorize FDA to require “certain research into drugs used in pediatric patients.” In fact, FDA has routinely enforced Public Law 108–155 known as the Pediatric Research Equity Act (PREA). Unless a waiver is granted, FDA almost always requires PK studies “to support dosing and administration for each pediatric subpopulation for which the drug or the biological product is safe and effective.”[62]
11. GERIATRIC CONSIDERATIONS
Because most PK studies are conducted in healthy volunteers between the ages of 18 and 55 years, there are important physiological patient aspects that should be considered for young versus older adults, often related to impairment of excretory functions. Determining if there are differences in the pharmacokinetic expression of the drug in elderly subjects and younger adults is critical. Likewise it is important to characterize those differences, as well as effects of influences, such as abnormal renal or hepatic function, that are more common in the elderly yet may occur in any age group. A sponsor may garner information regarding age-related differences in the pharmacokinetics of a drug, from a PK screening (that includes all ages, which may rule out a formal PK study) or from formal pharmacokinetic studies in the elderly and specifically in patients with excretory impairment.[63]
Sponsors should narrow the specialized elderly PK testing to those drugs that would have predominant use in this patient population, i.e. drugs used to treat Alzheimer’s and hypertension. Study populations should be focused on patients 65 and older with no upper limits, as this would be the primary demographic for these types of medications.[64]
It is not easy to recruit older geriatric volunteers for these special PK studies to compare to younger volunteers. Simply put, adverse drug events and drug-drug interactions occur more frequently in the oldest of patients, in part because they are more likely to be using complex drug therapies. Thus, willing participants tend to be younger, healthier geriatrics, not the very old (e.g., over the age of 85) or frail. Therefore, extrapolating dosages and possible adverse effects from either geriatric sub-population to the other may or may not be appropriate to yield the information sought.[65]
Moreover, as people age, various physiologic factors contribute to increased inter-individual variability in terms of ADME parameters. Cardiac structure/function, renal/GI systems and body composition are among the systems most often implicated in the PK/PD differences between old and young.
The following is a table of age-related physiological changes with ageing that may affect PK.[66]
Table 3. Physiologic Changes with Aging that May Affect Pharmacokinetics
[wpsm_comparison_table id=”24″ class=”customTable”]
(a) A chemical (e.g., cyclic AMP or Ca++) inside the postsynaptic neuron released by a first messenger (e.g., a neurotransmitter) and responsible for downstream regulation of a pathway or gene expression.
Adapted from Therapeutic Drug Monitoring in the Geriatric Patient, Susan W. Miller.
CONCLUSION
Proper design and placement of your PK studies is critical. Quite simply (using the case of ANDAs), the regulations require the applicant to use “the most accurate, sensitive, and reproducible approach available among those set forth.”[67]
While fiscal year 2016 saw a record 835 ANDA approvals (includes 184 tentative approvals) there were also 1,725 complete response letters (CRL).[68]
Many of us recall the incident during the first decade of the 21st century when, after several inspections of a well-known CRO, FDA called into question PK data spanning a four-year period, including multiple approvals. In doing so, FDA issued over a thousand notifications to companies with both pending and approved applications, noting that significant concerns about the validity of the results of the studies in support of marketing applications existed.[69]
As such, options offered by FDA included, repeat of the BE studies, re-assay of study samples at a different bioanalytical facility, or scientific audit by a qualified independent expert, knowledgeable in the area of BE studies and bioanalytical data.[70]
PDG® conducted a number of those scientific audits and has designed a multitude of BA/BE studies for a variety of applications. We routinely source, qualify and oversee the activities of CROs for our clients. When selecting a CRO, there are many factors we consider, and they always depend on the nature of the trial to be conducted. Regardless of the trial, we always seek and validate a solid track record, sound processes, up-to-date technology, compliant quality systems, and willingness and ability to adhere to clear performance parameters. Contact us for help with your next project.
Jodi Hutchins is an Independent Regulatory and Quality Consultant with over 15 years of global medical device registration experience, to include FDA 510(k) submissions. She held her most recent position for 9 years, as QA/RA Director for a worldwide distributor of medical devices.
Charles Jaap is Vice-President of Operations and Business Development for PDG®, a global pharmaceutical and medical device consultant with extensive experience in the strategic development of drug products and medical devices.
The opinions and statements in this paper are solely those of Charles Jaap and Jodi Hutchins and do not necessarily reflect those of PDG®.
Sources:
[1] The ABCD of clinical pharmacokinetics; Matthew P. Doogue, Therapeutic Advances in Drug Safety, 2013.
[2] Pharmacokinetics Basics- Absorption, Distribution, Metabolism and Excretion; Sunalim, Pharmaxchange, April 9, 2011.
[3] Pharmacokinetics: The Absorption, Distribution, and Excretion of Drugs; Joy Sakai, Practical Pharmacology for the Pharmacy Technician, June 20, 2008.
[4] Area under the plasma drug concentration-time curve; Thierry Buclin, Marie Nicod, and Stephan Kellenberger, Pharmacokinetics, July 2009.
[5] Clinical pharmacokinetics chapter 1; Evan J. Begg, Instant Clinical Pharmacology, 2003.
[6] Clinical Pharmacology 1: Phase 1 Studies and Early Drug Development; Gerlie Gieser, FDA Office of Clinical Pharmacology.
[7] Ibid@6.
[8] Ibid@6.
[9] Pharmacodynamics: How Drugs Work; J K Aronson, The Hitchhiker’s Guide to Clinical Pharmacology, 2016.
[10] Ibid@6.
[11] PK/PD modelling and simulations: utility in drug development; Iris Rajman, Drug Discovery Today, 2008.
[12] Ibid@6.
[13] Ibid@11.
[14] Draft Guidance for Industry: Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA; December 2013.
[15] Draft Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products – General Considerations; July 2002.
[16] 21 U.S.C. 355(j)(2)(A).
[17] 21 CFR 314.94.
[18] Draft Guidance for Industry: Applications Covered by Section 505(b)(2); October 1999.
[19] Guidance for Industry: Bioequivalence Recommendations for Specific Products; June 2010.
[20] Ibid@19.
[21] Ibid@19.
[22] FDA Adds 67 New/Revised Product Specific Bioequivalence Guidances; Pharmaceutical Development Group Breaking News, as accessed at www.pharmdevgroup.com on January 16, 2017.
[23] 21 CFR 320.22.
[24] Ibid@23.
[25] Ibid@23.
[26] Ibid@23.
[27] Ibid@23.
[28] Draft Guidance for Industry: Waiver of in Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System, May 2015.
[29] Biologics and Biosimilars: Background and Key Issues, Judith A. Johnson, Specialist in Biomedical Policy, September 7, 2016, Congressional Research Service Report 7-5700 RL44620 Prepared for Members and Committees of Congress.
[30] Information for Industry (Biosimilars); as accessed at www.fda.gov on January 16, 2017, page last updated January 12, 2017.
[31] Biosimilars in the US: Progress and Promise; John Jenkins, Director Office of New Drugs, October 27, 2016.
[32] Ibid@30.
[33] Ibid@29.
[34] ICH Guidance Tripartite Guideline: Ethnic Factors in the Acceptability of Foreign Clinical Data E5(R1); February 1998.
[35] Epoetin alfa Package Insert; April 1993.
[36] Clinical efficacy and safety of HX575, a biosimilar recombinant human erythropoietin, in the management of anemia; Ivo Abraham and Karen MacDonald, Biosimilars, 2012.
[37] Comparison of the Pharmacokinetic and Pharmacodynamic Profiles of One US-Marketed and Two European-Marketed Epoetin Alfas. A Randomized Prospective Study; Lissy Michael, et. al., Drugs R D, 2011.
[38] Narcan® nasal spray Summary Review; November 18, 2015.
[39] Ibid@38.
[40] Guidance for Industry: Population Pharmacokinetics; February 1999.
[41] Ibid@40.
[42] A population approach to characterise amisulpride pharmacokinetics in older people and Alzheimer’s disease; Suzanne Reeves, et. al., Psychopharmacology, 2016.
[43] Dose-Response Relationships; Abimbola Farinde, Merck Manual Professional Version, October 2016.
[44] Phase 1 Randomized, Double-Blind, Placebo-Controlled Study to Determine the Safety, Tolerability, and Pharmacokinetics of a Single Escalating Dose and Repeated Doses of CN-105 in Healthy Adult Subjects; JT Guptill, et. al., The Journal of Clinical Pharmacology, 2016.
[45] Ibid@18.
[46] Changing dosing regimen could improve efficacy of malaria vaccine candidate, study shows; News Medical Life Sciences, September 7, 2016.
[47] Drugs@FDA Glossary of Terms; as accessed at www.fda.gov on January 16, 2017, Page last updated February 2, 2012.
[48] Route of Administration; FDA Data Standards Manual (monographs), Vol. 004, January 11, 2006.
[49] Drug Administration; Debra Si Mui Sim, Pharmacological Basis of Acute Care, 2015.
[50] Ibid@49.
[51] Pharmacodynamics & Pharmacokinetics 1; Dr. Jennifer Shabbits, September 9, 2014.
[52] The Influence of Administration Route in the Comparison of Dosage Forms; Tansel Comoglu, Archives of Trauma Research, 2013.
[53] Ibid@49.
[54] Guidance for Industry: Food-Effect Bioavailability and Fed Bioequivalence Studies; December 2002.
[55] Draft Guidance for Industry: Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA; December 2013.
[56] Draft Guidance for Industry: Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs General Considerations; March 2014.
[57] Ibid@54.
[58] Ibid@54.
[59] Factors and Mechanisms for Pharmacokinetic Differences between Pediatric Population and Adults; Eva Fernandez, Pharmaceutics, 2011.
[60] Children are not Little Adults; WHO Training Package for the Health Sector. July 2008.
[61] Ibid@59.
[62] Pediatric Research Equity Act of 2003; Public Law 108-155, December 3, 2003.
[63] ICH Topic E 7: Studies in Support of Special Populations: Geriatrics; European Medicines Agency, March 1994.
[64] Ibid@63.
[65] Therapeutic Drug Monitoring in the Geriatric Patient; Susan W. Miller, Clinical Pharmacokinetics, 2012.
[66] Ibid@65.
[67] Ibid@14.
[68] FY 2016 Generic Approvals by FDA: A New Record Under GDUFA; Zachary Brennan, Regulatory Affairs Professionals Society, Regulatory Focus, October 12, 2016.
[69] MDS hit by FDA warning on pharmacokinetic data; PharmaTimes Online, January 15, 2007.
[70] Full text of FDA Letter to Pending ANDA Holders Warning About MDS; as accessed at www.archive.org on January 16, 2017, Internet Archive, January 10, 2007.
![]()





